- 移动端


上海和元生物技术(集团)股份有限公司品牌商
14 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5

推荐产品
公司新闻/正文
Nat Cardiovasc Res|揭示CD248+成纤维细胞介导的成纤维-免疫交互在心脏纤维化中的重要作用
475 人阅读发布时间:2025-09-17 10:26
心肌梗死(Myocardial infarction, MI)后心脏不良重塑是心力衰竭发生发展的重要病理基础,心脏纤维化是该过程中的关键驱动因素,已成为制约心血管疾病治疗的瓶颈。精准解析心梗后不同区域中成纤维细胞亚群的动态变化,进而寻找特异在心梗中晚期病理重塑中起关键作用的成纤维细胞亚群,以期在维持早期修复功能的同时抑制纤维化恶性进展,或将成为突破当前纤维化治疗困境的关键路径。
2025年3月27日,浙江大学胡新央团队在Nature Cardiovascular Research发表了题为“Dynamic molecular atlas of cardiac fibrosis at single-cell resolution shows CD248 in cardiac fibroblasts orchestrates interactions with immune cells”的研究论文。本研究通过整合单细胞RNA测序、空间转录组测序,系统解析了成纤维细胞时空异质性在心梗病理重塑中的调控作用,发现CD248+成纤维细胞是一群特异参与心梗中晚期心脏不良重塑的成纤维细胞亚群,其机制与其促进心脏T细胞浸润进而介导免疫-成纤维交互作用相关。利用单抗及嵌合抗原受体(chimeric antigen receptor, CAR)技术特异干预该群成纤维细胞可减轻心脏纤维化并改善心脏功能。
研究结果
01 CD248+成纤维细胞是心肌梗死后慢性纤维化的关键亚群
为了探究心肌梗死后心肌成纤维细胞的细胞图谱,收集心肌梗死3天和28天后小鼠心肌组织PDGFR-α和DDR2双阳性成纤维细胞进行单细胞RNA测序(scRNA-seq)。获得了69,705个心脏成纤维细胞(图1a)。这13个亚群代表性基因分别是Mt1/Tcf21 (F1)、Smoc2/Gsn (F2)、Upk3b/Msln (F3)、Ccl2/Ptx3 (F4)、Saa3/Serpinb2 (F5)、Apoe/Fmo2 (F6)、Thbs4/Postn (F7)、Comp/ frp2 (F8)、Cd248/Ackr3 (F9)、Cthrc1/Ccl9 (F10)、Lars2/Hspg2 (F11)、Acta2/Timp1 (F12)和Atf3/Fosb (F13) (图1 b)。有趣的是,F9亚群(Cd248/Ackr3)在第28天比例明显增加(图1 c)。GO分析表明F9参与ECM和细胞粘附,提示F9与胶原合成和炎症有很强的相关性(图1d)。心肌梗死28天后CD248表达升高,且在梗死区富集(图1f)。在心肌梗死患者心脏组织单细胞RNA数据中,CD248主要在hF5亚群中高表达(图g、h)。
小鼠心脏的空间转录组(Stereo-seq)显示,心肌梗死后14天和28天,CD248成纤维细胞数显著增加,且在梗死区和梗死周围区富集(图1j,k)。人梗死心脏组织的空间转录组分析显示,纤维化区域内的CD248成纤维细胞显著增加(图1l,m)。这些发现强调了CD248+成纤维细胞在心脏损伤后长期纤维化重塑中的潜在作用。通过流式进一步证实心肌梗死相关成纤维细胞中CD248上调(图1n,o)。心肌组织中CD248在14天时明显升高,并在心肌梗死后28天保持持续升高(图1q,r)。综上所述,确定了心肌梗死后特异性诱导的心脏成纤维细胞亚群(CD248+成纤维细胞)。
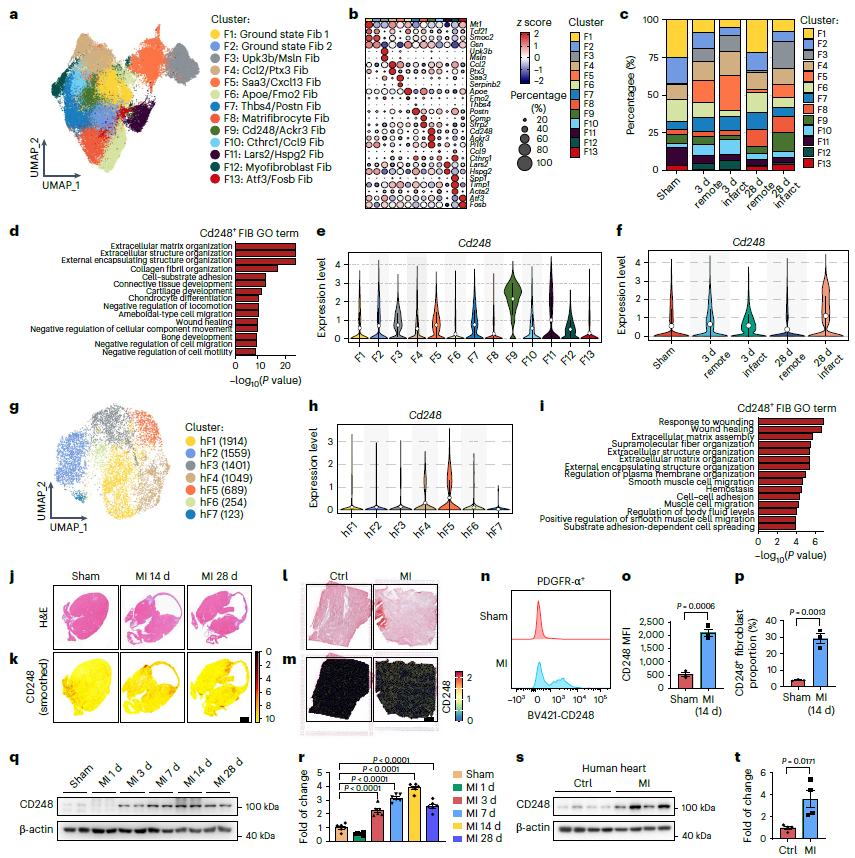
图1 CD248+成纤维细胞是心肌梗死后慢性纤维化的关键亚群
02 CD248缺失可减轻缺血条件下的心脏纤维化和功能障碍
为了阐明CD248在心脏纤维化中的作用,构建了CD248敲除小鼠(图2a)。与WT小鼠相比,CD248 KO小鼠的心功能明显变化 (图2a),心脏纤维化面积明显减少(图2b,c)。I/R后,CD248 KO心脏中纤维化相关蛋白也显著降低(图2d,e)。更有趣的是,CD248失活显著减轻了I/R损伤小鼠心脏的炎症,降低了Il1b, Il6和Il33 mRNA水平(图2f)。考虑到基质纤维细胞中CD248的表达相对较低,这种减少可能是由于CD248+成纤维细胞的缺失。在I/R后28天,CD248Postn-CreER小鼠的心功能得到了显著改善,纤维化面积也显著减少(图2j、l)。总之,这些结果证实了CD248在促进损伤后慢性心脏纤维化和心脏炎症中具有不可或缺的作用。
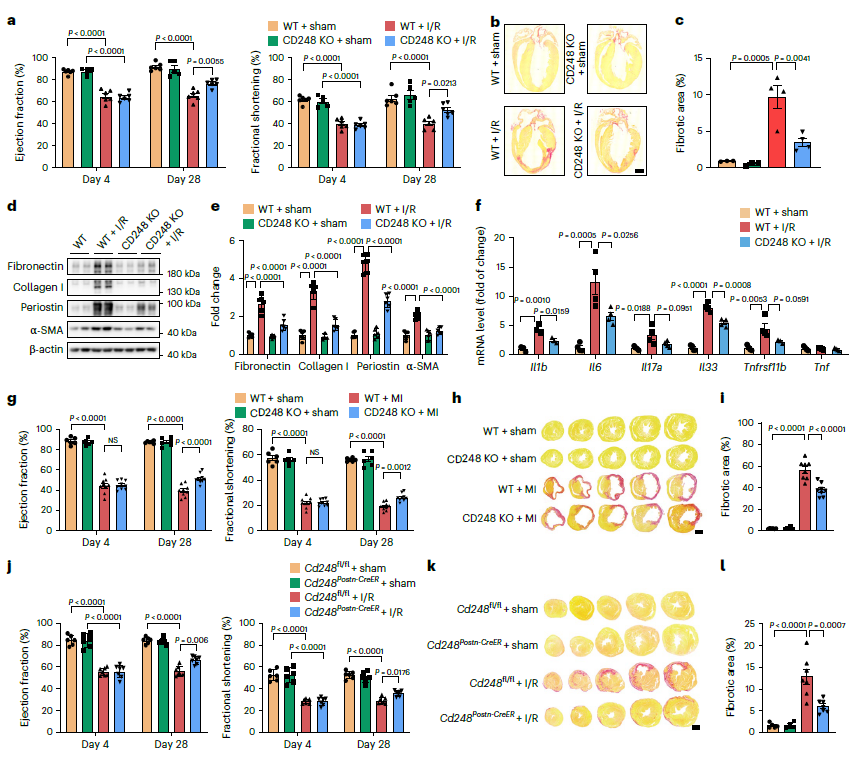
图2 CD248缺失可减轻缺血条件下的心脏纤维化和功能障碍
03 CD248成纤维细胞促进T细胞浸润,引发成纤维细胞与T细胞相互作用
对心肌梗死后14天小鼠心脏的CD248+和CD248-成纤维细胞进行RNA测序。GO分析显示,细胞迁移、细胞间粘、ECM等在CD248+成纤维细胞中显著性富集(图3a)。KEGG分析表明,CD248+成纤维细胞参与炎症调节、趋化因子信号传导、IL-17信号传导和TH 17细胞分化(图3b),表明CD248+成纤维细胞参与缺血性损伤后的炎症微环境调节。在CD248 KO小鼠心脏中,I/R后第7天T细胞浸润明显减少(图3c)。更具体地说,CD248失活减少了CD4 T细胞的浸润,特别是TH1和TH17亚群(图3d)。心肌梗死小鼠模型也重现了CD248失活导致的心肌CD4 T细胞浸润减少(图3e)。简而言之,CD248+成纤维细胞特异性地调节了T细胞在损伤心肌中的慢性浸润。
利用心肌梗死后14天和28天小鼠心脏组织的空间转录组图谱,绘制了CD248和CD3在心肌中的分布模式,并发现它们在成纤维细胞中的免疫生态位(CD248/CD3;图3f)。在梗死的人类心脏组织中重现了相同的分布模式(图3j)。使用SPOTlight对每个点的相对细胞组成进行反卷积,并计算细胞间的相关性。邻域相关分析表明,CD248+成纤维细胞与成纤维细胞免疫生态位中的T细胞有很强的相关性(图3g)。对CD248阳性/阴性成纤维细胞与T细胞之间的距离进行量化,再次证实CD248+成纤维细胞与T细胞之间的距离比CD248−成纤维细胞与T细胞之间的距离短(图3i)。在梗死的人类心脏中也发现了CD248+成纤维细胞和T细胞之间类似的相互作用模式(图3k、1)。综上所述,CD248+成纤维细胞促进T细胞浸润,引发成纤维细胞与T细胞相互作用,从而加剧缺血心脏组织的炎症微环境。
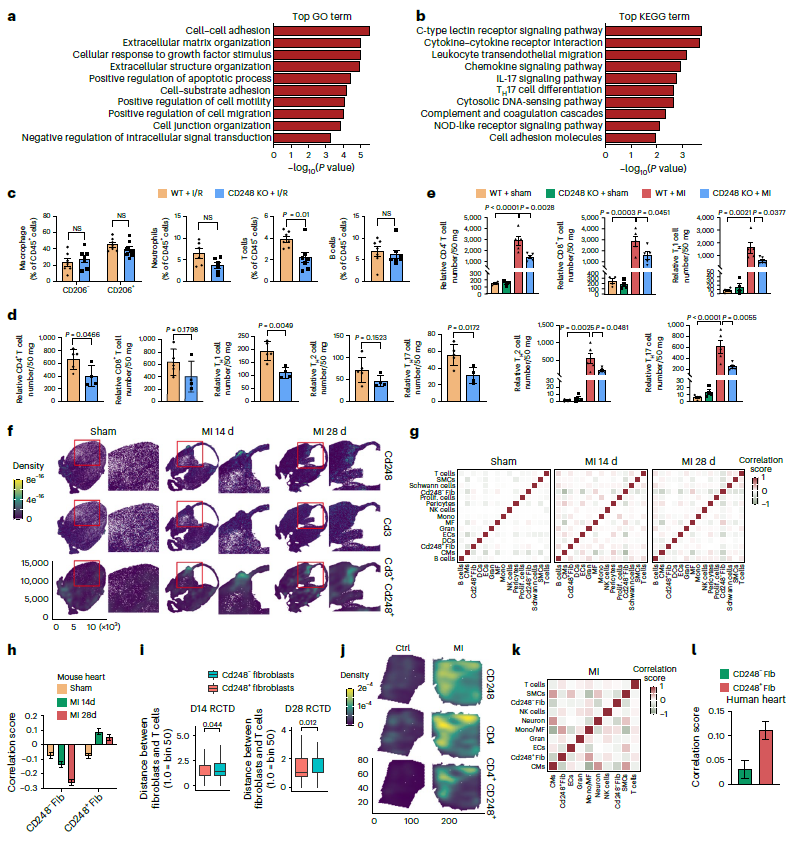
图3 CD248+成纤维细胞促进T细胞浸润,引发成纤维细胞与T细胞相互作用
04 ACKR3介导了CD248+成纤维细胞对T细胞的浸润增加
在Cd248+成纤维细胞中的前十趋化因子和受体中,Ackr3及CXCR7特异性高表达(图4a)。在蛋白水平上也观察到ACKR3在CD248+成纤维细胞中的诱导作用(图4b,c)。I/R损伤后第7天,CD248失活显著降低了心脏成纤维细胞中ACKR3的表达(图4d,e)。通过对新生小鼠心脏成纤维细胞(nmcf)和CD4 T细胞的体外共培养实验(图4f,g),发现nmcf中CD248过表达显著增强T细胞迁移。沉默Ackr3显著抑制了CD248 过表达诱导的T细胞迁移(图4f,g),表明Ackr3在CD248+成纤维细胞介导的T细胞迁移中起关键作用。
为了证明成纤维细胞ACKR3在CD248+成纤维细胞与T细胞互作中的作用,用CD248 过表达慢病毒和ACKR3敲低慢病毒在心脏I/R小鼠模型中进行了体内实验 (图4 h, j,k)。成纤维细胞特异性CD248 过表达加重了I/ r诱导的心功能障碍和纤维化(图4i, l, m)。相反,成纤维细胞特异性ACKR3敲低挽救了CD248 过表达诱导的心功能障碍和纤维化加重(图4i、l、m)。更有趣的是,成纤维细胞特异性CD248过表达促进了I/R损伤小鼠心脏中CD4 T细胞的浸润,这一过程被成纤维细胞特异性ACKR3敲低所逆转(图4n)。综上表明,CD248+成纤维细胞对T细胞的浸润增加是由ACKR3介导的。
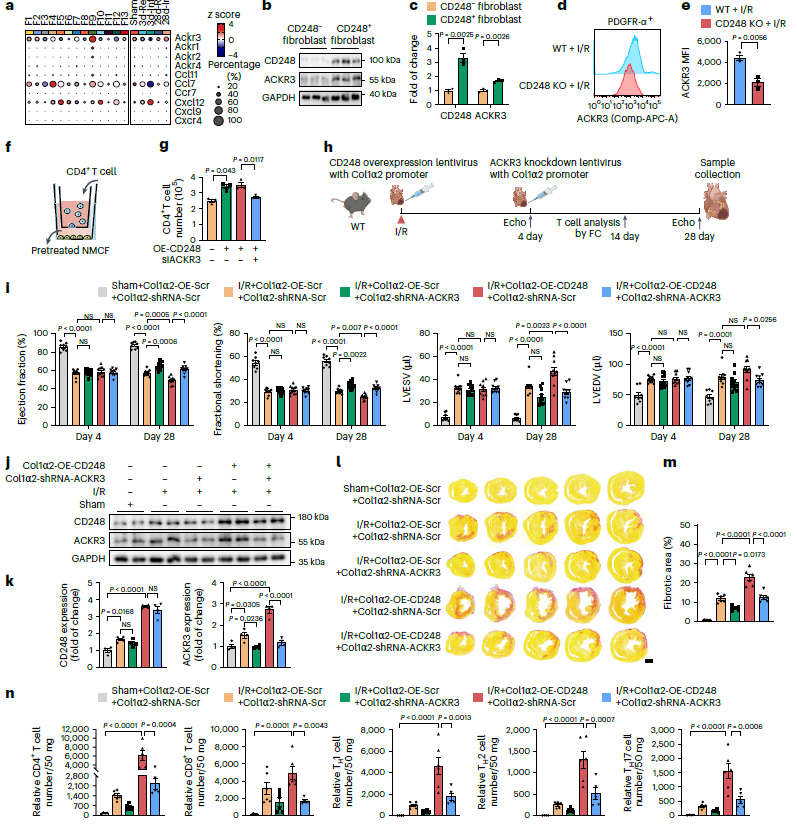
图4 ACKR3介导了CD248+成纤维细胞对T细胞的浸润增加
05 CD248通过稳定TGFβRI抑制ACKR3降解
为了阐明CD248和ACKR3之间的潜在关系,进行了CD248 过表达和敲低实验来观察ACKR3在nmcf中的改变。蛋白酶抑制剂MG132减弱了ACKR3蛋白的下调(图5a,b),这意味着CD248失活引发ACKR3蛋白下调的过程中存在蛋白酶依赖性降解。敲除CD248会增加ACKR3的泛素化水平(图5c)。
鉴于TGF-β信号在ECM重塑和免疫调节中的重要作用,进一步研究了TGF-β信号在CD248对ACKR3的影响中的潜在作用。在CD248 KO小鼠心脏和CD248过表达的nmcf中的进一步研究表明,CD248调控I型和II型转化生长因子β受体(TGFβRI和TGFβRII)的表达(图5d,e)。同时,Cd248敲低在培养的nmcf中下调TGFβRI的表达;溶酶体抑制剂BafA1可以阻断这种作用,而蛋白酶体抑制剂则不能。因此推测CD248与TGFβRI的结合抑制了TGFβRI的溶酶体降解。
进一步研究了TGFβRI在CD248调节的ACKR3稳定和T细胞迁移中的潜在作用。抑制TGFβRI可显著降低nmcf中ACKR3的表达(图5f,g)。此外,CD248敲低诱导的ACKR3下调被TGFβRI 过表达逆转,表明TGFβRI参与了CD248对ACKR3的调节(图5h-j)。此外,抑制TGFβRI可降低CD248过表达诱导的T细胞迁移,表明TGFβRI在CD248促进的T细胞迁移中起重要作用(图5k, l)。为了阐明TGFβRI调控ACKR3的机制,再次用溶酶体抑制剂巴菲霉素或蛋白酶体抑制剂MG132孵育经TGFβRI或si-TGFβRI处理的nmcf,发现蛋白酶体抑制剂能够阻止TGFβRI下调诱导的ACKR3下调(图5m,n)。总之,CD248通过抑制其溶酶体降解来稳定TGFβRI,进而激活TGFβ信号传导并抑制ACKR3降解,从而促进成纤维细胞与T细胞的相互作用。
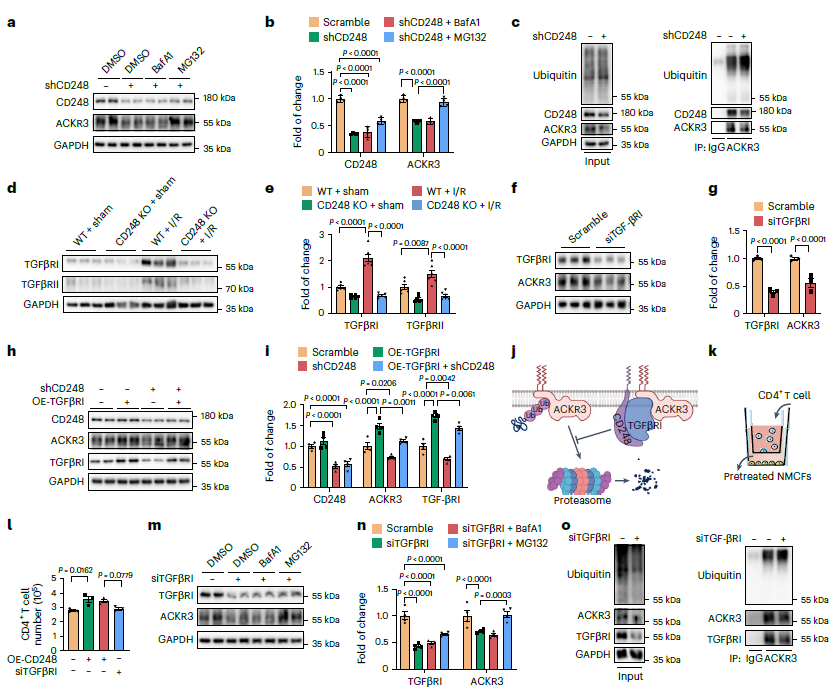
图5 CD248通过稳定TGFβRI抑制ACKR3降解
06 CD248单克隆抗体可改善I/R后心脏纤维化和功能障碍
为探讨靶向干预策略,制备了靶向CD248的单克隆抗体IgG78,该单克隆抗体与CD248具有高亲和力和特异性结合,并以剂量依赖的方式结合。为了证实其体内治疗潜力,在小鼠I/R损伤后,每周两次静脉注射IgG78(图6a)。IgG78治疗可显著改善心功能,改善EF、FS,降低LVEDV和LVESV(图6b)。在I/R损伤的心脏组织中,IgG78也显著降低了纤维化和纤维化相关蛋白的表达(图6c-f)。更有趣的是,I/R后IgG78处理显著降低了CD4 T细胞浸润和炎症细胞因子表达(图6g,h)。综上所述,CD248单克隆抗体阻断CD248介导的成纤维细胞-T细胞相互作用,可预防I/R诱导的心脏纤维化和功能障碍。
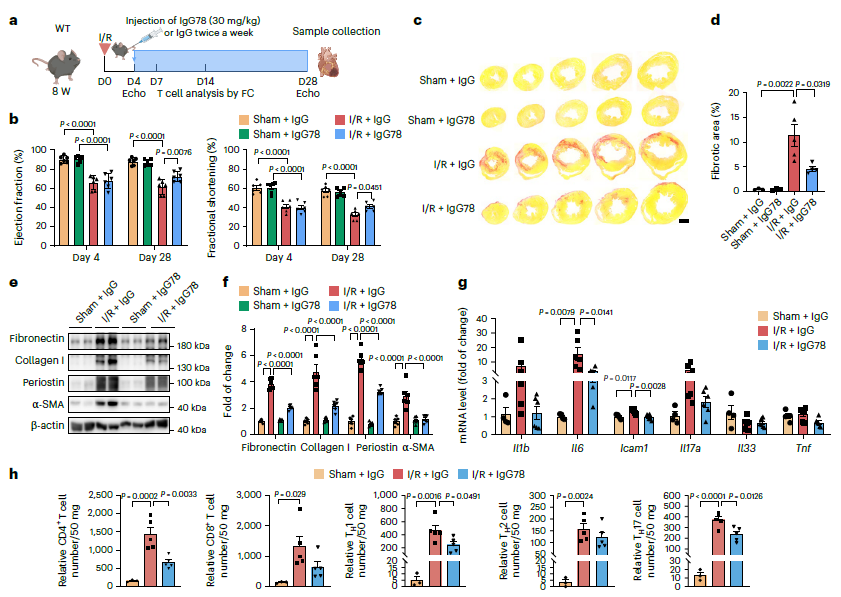
图6 CD248单克隆抗体可改善I/R后心脏纤维化和功能障碍
07 CD248 CAR-T细胞可减轻I/R后的心肌纤维化和功能障碍
为了探索CD248 CAR-T治疗缺血性心脏病的疗效,基于IgG78的Fab片段构建了一种CD248特异性CAR。CD248 CAR-T细胞治疗可改善心脏功能(图7b)。CD248 CAR-T组的纤维化面积也比PSCA CAR-T细胞组显著减少(图7c,d)。在I/R后28天,与PSCA CAR-T相比,CD248 CAR-T细胞组的CD248成纤维细胞显著减少(图7e,f),进一步证明CD248 CAR-T细胞有效地消除了CD248成纤维细胞,也能显著减少I/R损伤小鼠心脏中CD4和CD8 T细胞的浸润(图7g)。总之,CD248 CAR-T细胞主要通过消除缺血区域的CD248+成纤维细胞,来达到减少纤维化和改善心功能的作用。
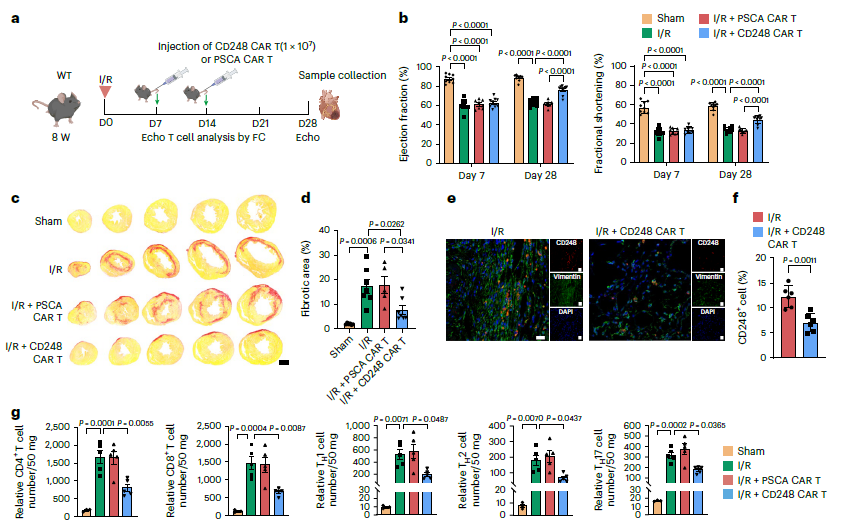
图7 CD248 CAR-T细胞可减轻I/R后的心肌纤维化和功能障碍
08 IgG78和CD248 CAR-T细胞治疗在I/R后发挥抗纤维化作用
为了探索IgG78和CAR-T细胞治疗是否可以在I/R后的慢性阶段停止纤维化区域扩张或逆转纤维化,从I/R后28天开始,每周静脉注射两次IgG78,持续6周(图8a)。在I/R后70天,IgG78治疗组表现出心功能明显改善(图8b,c),并伴有纤维化面积减少(图8d,e)。迟发性IgG78治疗也减少了缺血心脏中CD3 T细胞的浸润(图8f,g)。同时,也评估了CD248 CAR-T细胞疗法的治疗效果。在I/R后14天和28天分别输注两次CD248 CAR-T细胞(图8)。CD248 CAR-T细胞治疗组的心功能有显著改善(图8i,j)。此外,CD248 CAR-T细胞治疗也显著减少了纤维化面积(图8k,1),并减少了CD3 T细胞浸润(图8m,n)。综上所述,在I/R后慢性期,利用IgG78和CD248 CAR-T细胞可阻断CD248+成纤维细胞与T细胞的相互作用,并对损伤心脏具有抗纤维化作用。
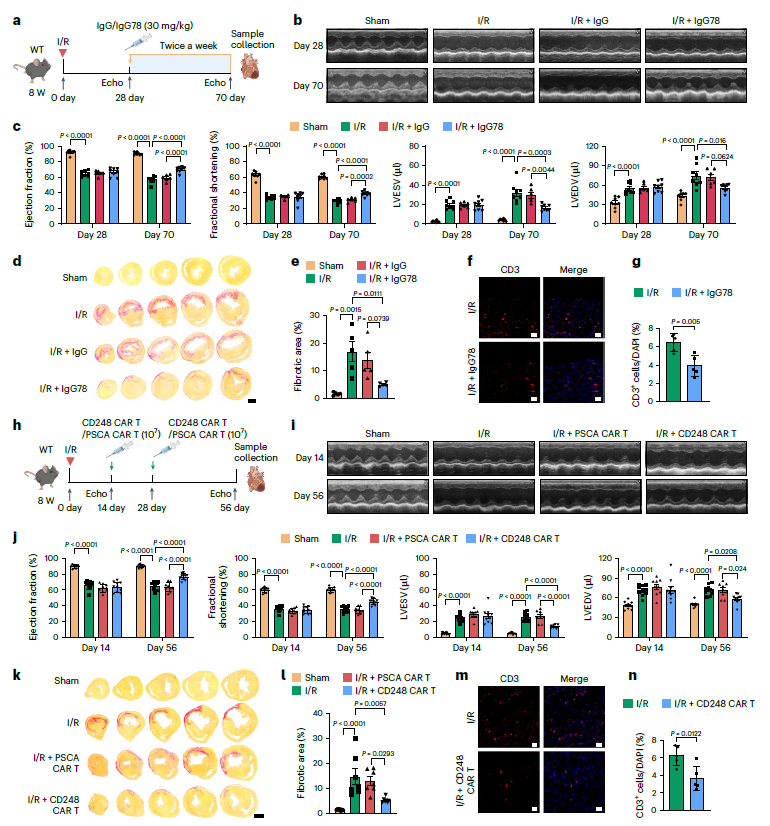
图8 IgG78和CD248 CAR-T细胞治疗在I/R后发挥抗纤维化作用
和元服务
和元生物提供多组学服务(如全外显子测序、转录组、Astral蛋白组、非靶向代谢组、非靶向脂质组、靶向代谢组、16s/ITS/18s扩增子测序、宏基因组、Cut&Tag、MeRIP (m6A)-seq、全基因组甲基化测序、简化甲基化测序等)和单细胞及空间转录组服务(如10x单细胞转录组、10x单细胞转录组及TCR/BCR、墨卓单细胞转录组、10x CytAssist空间转录组、10x HD空间转录组、Stereo-seq 空间转录组等),致力于为广大生命科学家、医学工作者提供基于多组学的科研及临床应用解决方案。
业务咨询:15800353038

