- 移动端


上海和元生物技术(集团)股份有限公司品牌商
14 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5

推荐产品
公司新闻/正文
【肺癌研究文章集锦】肺癌耐药与免疫治疗困境破局--多维前沿靶点开辟治疗新路径
523 人阅读发布时间:2025-09-25 16:27
肺癌(lung cancer),也称支气管肺癌,是全球常见的癌症之一,具有高发病率和死亡率。根据癌细胞组织学,肺癌主要分为非小细胞肺癌(NSCLC)和小细胞肺癌(SCLC)两种类型。非小细胞肺癌(NSCLC)是最常见的类型,占所有肺癌的85%左右。NSCLC中约 40%为腺癌,25%至30%为鳞状细胞癌,10%至15%为大细胞癌。SCLC是一种极为恶性的癌症,与 NSCLC 相比,SCLC 通常更具侵袭性,生长速度更快。SCLC具有早转移倾向,确诊时常已发生转移,容易侵及淋巴结、脑部、肝脏等远端部位,预后差。
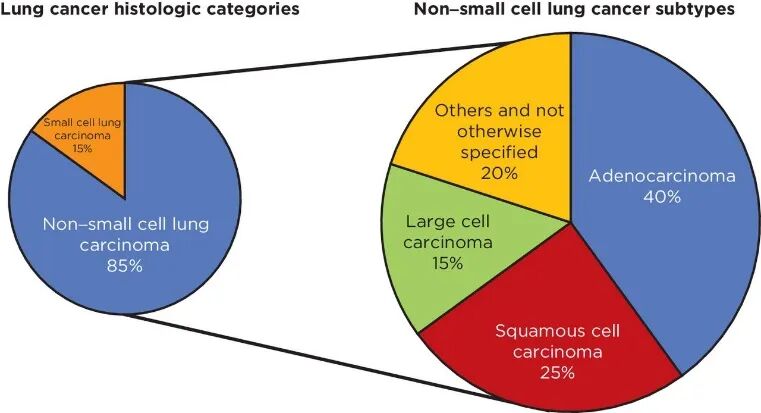
图1. 肺癌的组织学分类
肺癌的分子发病机制过程相当复杂且异质性,它是一个多步骤、多因素的复杂过程,其核心是一场发生在细胞内部的“通路之争”。在这场战争中,两大主角决定着细胞的命运:
“加速器”(原癌基因通路)
如EGFR, KRAS等基因发生突变,使其功能“失控”,持续向细胞发出“分裂增殖”的指令,让细胞疯狂生长。
“刹车片”(肿瘤抑制通路)
如TP53, RB1等基因失活或缺失,导致本应负责“终止分裂”、“修复错误”或“启动凋亡”的机制失灵。
当“加速器”被持续踩下,而“刹车片”又被拆除时,细胞便脱离了正常调控,走向癌变。我们如今的治疗策略(如靶向治疗),正是基于对这些分子通路的深刻理解,旨在精准地修复“刹车”或抑制“油门”,为患者带来生的希望。当然除了这两个主角色,其他因素也起到非常重要的影响,比如DNA 修复过程紊乱、生长因子和血管生成表达的增加、细胞周期调节异常、抗凋亡和促凋亡基因的突变等等……
分子靶向治疗已成为非小细胞肺癌(NSCLC)的关键治疗策略。多种受体酪氨酸激酶参与细胞生长和存活,如表皮生长因子受体(EGFR)、肝细胞生长因子受体(c-Met)、间变性大细胞淋巴瘤激酶(ALK)等,这些激酶是分子靶向治疗的重要靶点。C-ROS原癌基因1酪氨酸激酶(ROS1)、鼠类肉瘤病毒癌基因(KRAS)、鼠类肉瘤病毒癌基因同源物B1(BRAF)、间质–上皮细胞转化因子(MET)、转导重排基因(RET)等靶点治疗也是目前研究重点。以 EGFR突变作为治疗靶点,已开发出吉非替尼、厄洛替尼和奥希替尼等酪氨酸激酶抑制剂。针对ALK和ROS1基因重排的患者,开发出克唑替尼,用于治疗ALK阳性NSCLC,后续研发了包括阿来替尼、布加替尼、塞普替尼等多种针对ALK的药物,用于克服克唑替尼的耐药。此外,针对其他靶点的抑制剂也在持续研究中。
尽管分子靶向治疗策略最初有效,但由于表观遗传改变和肿瘤异质性,获得性耐药通常不可避免,这严重限制了 NSCLC的治疗效果。
面对这一严峻挑战,寻找新的作用靶点和治疗方案已成为领域内的迫切需求。我们深感荣幸,通过可靠的病毒工具与技术服务,助力多位科研伙伴在这一前沿方向取得突破。以下为大家分享部分精彩客户文献:
研究一
可变多聚腺苷酸化遗传调控为非小细胞肺癌分子机制提供新见解
文章标题:Genetic Regulation of Alternative Polyadenylation Provides Novel Insights into Molecular Mechanisms Underlying Non‐small Cell Lung Cancer
发表期刊:Advanced Science(IF 14.1)
研究团队:华中科技大学同济医学院
研究背景:可变多聚腺苷酸化(APA)是转录后调控的重要机制,影响mRNA的3'UTR长度,进而调控基因表达。虽然APA在多种癌症中异常活跃,但其遗传调控在NSCLC中的具体作用尚未系统研究。
结果:研究团队利用TCGA中1010例NSCLC样本的基因型和APA数据,进行了全基因组apaQTL(APA相关的遗传变异)分析,识别出117,161个显著apaQTL变异,其富集于3’UTR、poly(A)信号、RBP结合区域,且独立于eQTLs(表达数量性状位点);通过将 apaQTLs 与 NSCLC GWAS 数据整合分析,发现 apaQTLs 在 NSCLC GWAS 位点中显著富集,证明整合 apaQTLs 可提升 NSCLC 风险预测准确性。随后通过整合 apaQTLs 与 OncoArray Consortium 的 NSCLC GWAS 数据,筛选出与 NSCLC 风险显著相关的 apaQTL 变异 rs9606(位于 LYRM4 基因 3'UTR)。接着通过3’RACE、双荧光素酶报告系统、RNA pull-down和RIP等实验证明rs9606-T allele导致LYRM4转录本3’UTR缩短,从而增强LYRM4 mRNA稳定性和翻译效率。后续通过细胞表型和裸鼠成瘤实验证明LYRM4高表达可以促进NSCLC细胞增殖、迁移和肿瘤形成。最后通过Co-IP和铁死亡相关指标检测,发现LYRM4通过抑制铁死亡促进肿瘤发生,且rs9606-T allele抑制效果更强。
总结:本研究揭示了APA相关遗传变异在NSCLC中的重要作用,为理解NSCLC的发病机制提供了新视角。同时,rs9606的功能机制为NSCLC的精准预防和治疗提供了潜在靶点。结合apaQTLs的PRS模型有望成为NSCLC风险预测的新工具。
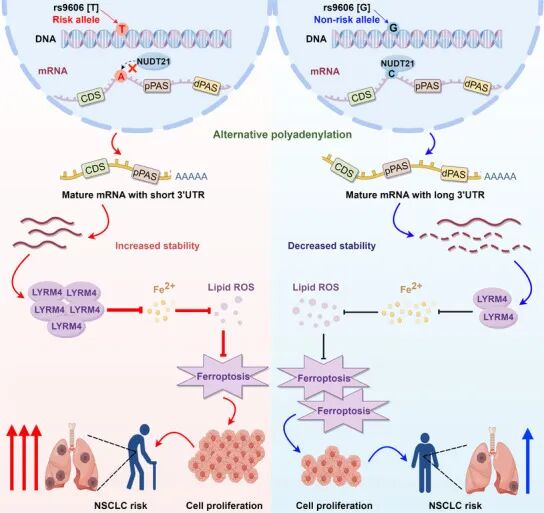
图2. 机制示意图
和元助力
和元生物有幸为客户提供LYRM4过表达慢病毒,用于感染A-549和NCI-H1299(人非小细胞肺癌细胞)
研究二
干扰素-γ刺激的抗原呈递癌症相关成纤维细胞阻碍肺癌新辅助化学免疫治疗疗效
文章标题:Interferon-γ-stimulated antigen-presenting cancer-associated fibroblasts hinder neoadjuvant chemoimmunotherapy efficacy in lung cancer
发表期刊:Cell Reports Medicine(IF 11.7)
研究团队:上海交通大学胸科医院
研究背景:新辅助化疗免疫疗法(NCIT)已成为可切除非小细胞肺癌(NSCLC)患者的标准治疗之一,通过在手术前激活全身抗肿瘤免疫来改善预后。然而,至少50%的患者对NCIT无效,其抵抗机制尚不明确。本研究旨在探索肿瘤微环境(TME)中哪些因素导致了NCIT抵抗,并寻找潜在的治疗靶点。
结果:研究团队首先通过对10例NSCLC患者NCIT治疗前后的肿瘤样本进行scRNA-seq分析,发现抗原呈递CAFs(apCAFs)在non-MPR患者治疗后显著富集。进一步通过DSP空间转录组技术,在6例患者组织切片中定位apCAFs,确认其在纤维化“热点”区域聚集,且与Tregs空间距离显著接近。为了验证apCAFs是否直接导致耐药,研究者构建小鼠共移植模型,将患者来源的apCAFs与肿瘤细胞一同移植至小鼠体内,结果显示:apCAFs仅在抗PD-1治疗背景下显著促进肿瘤生长,并抑制CD8+ T细胞功能,提示其是免疫治疗的“帮凶”。
接下来,研究者追问:是什么激活了apCAFs?通过体外3D培养模型(PDTF)和细胞因子筛查,发现抗PD-1治疗会刺激肿瘤微环境释放IFN-γ,进而激活apCAFs中的JAK1/2-STAT1信号通路,诱导其高表达PD-L2和MHCII类分子。进一步机制探索揭示,apCAFs通过PD-L2与Tregs表面的RGMB结合,促进一类具有强免疫抑制功能的FOXP1+ Tregs的扩增,从而压制抗肿瘤免疫响应。最后,研究者在小鼠模型和PDTF平台中证明,抗PD-1联合抗PD-L2或抗RGMB抗体治疗可显著逆转apCAFs介导的免疫治疗抵抗,恢复CD8+ T细胞的杀伤功能。
总结:该研究首次完整揭示了apCAFs通过IFNγ-JAK-STAT-PD-L2-RGMB信号轴驱动NCIT耐药的新机制,也为克服免疫治疗耐药提供了新的联合治疗策略(如抗PD-L2/RGMB疗法),具有重要的临床转化潜力。
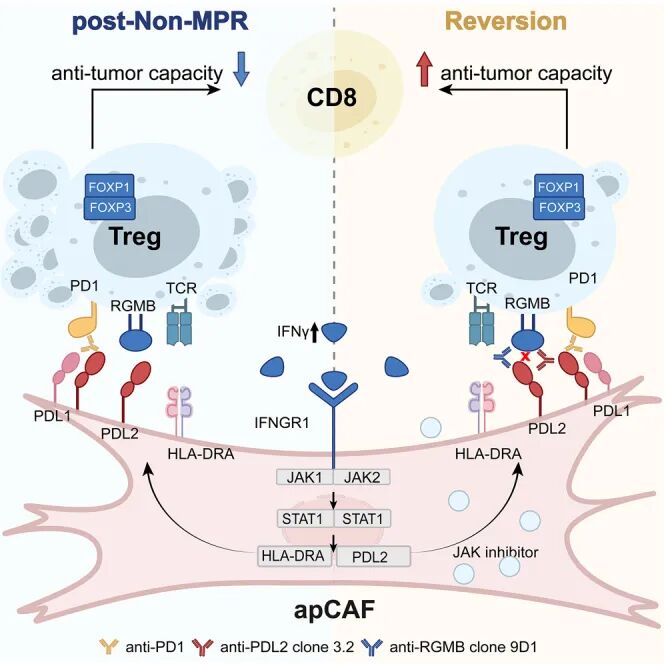
图3. 机制示意图
和元助力
和元生物有幸为客户提供si-IFI6,si-IFI27,用于感染CAFs(癌症相关成纤维细胞)
研究三
RAC1 剪接变异加速肿瘤发生并定义了肺癌的潜在治疗靶点
文章标题:Splicing Shift of RAC1 Accelerates Tumorigenesis and Defines a Potent Therapeutic Target in Lung Cancer
发表期刊:Advanced Science(IF 14.1)
研究团队:复旦大学基础医学院
研究背景:EGFR突变是肺腺癌(LUAD)的重要驱动因素,但耐药问题几乎不可避免。RNA剪接失调是肿瘤的新特征,RAC1基因通过可变剪接产生RAC1A和RAC1B两种异构体,但其在LUAD中的具体功能差异及调控机制尚不明确。本研究旨在揭示RAC1B的致癌作用及其作为治疗靶点的潜力。
结果:研究者首先通过TCGA和FUSCC队列的RNA-Seq数据发现,RAC1B在LUAD肿瘤组织中显著上调,尤其在EGFR突变患者中更为明显。生存分析显示高RAC1B表达与患者不良预后相关。为了明确RAC1B是否致癌,他们在EGFR/Trp53突变型小鼠肺内分别过表达RAC1A与RAC1B,发现只有RAC1B能显著加速肿瘤生长并缩短生存期。scRNA-seq分析进一步揭示RAC1B过表达组中恶性细胞和增殖细胞比例显著增加。为了验证RAC1B的必要性,研究者使用CRISPR-Cas9系统特异性敲低RAC1B,发现其能显著抑制肿瘤发展并延长小鼠生存。在人源LUAD细胞系(如PC9、H1975)中敲低RAC1B可抑制细胞增殖并诱导凋亡。RNA-Seq和功能富集分析表明RAC1B特异性调控RTK信号通路。进一步探究其机制发现,GEF蛋白GDS1特异性结合并激活RAC1B,抑制EGFR的溶酶体降解途径,延长其半衰期并增强下游ERK信号。最后研究者在PDO和PDX模型中验证了靶向RAC1B的治疗潜力,使用Dox诱导的shRNA或LNP包载的siRNA沉默RAC1B,可显著抑制肿瘤生长,并且与奥希替尼(Osi)联合治疗显示出显著更强的抑制作用。
总结:首次明确RAC1B(而非RAC1A)是EGFR突变型LUAD的关键促瘤因子,通过GDS1激活并稳定EGFR蛋白,提出靶向RAC1B的RNAi治疗策略,为克服EGFR-TKI耐药提供了新途径。
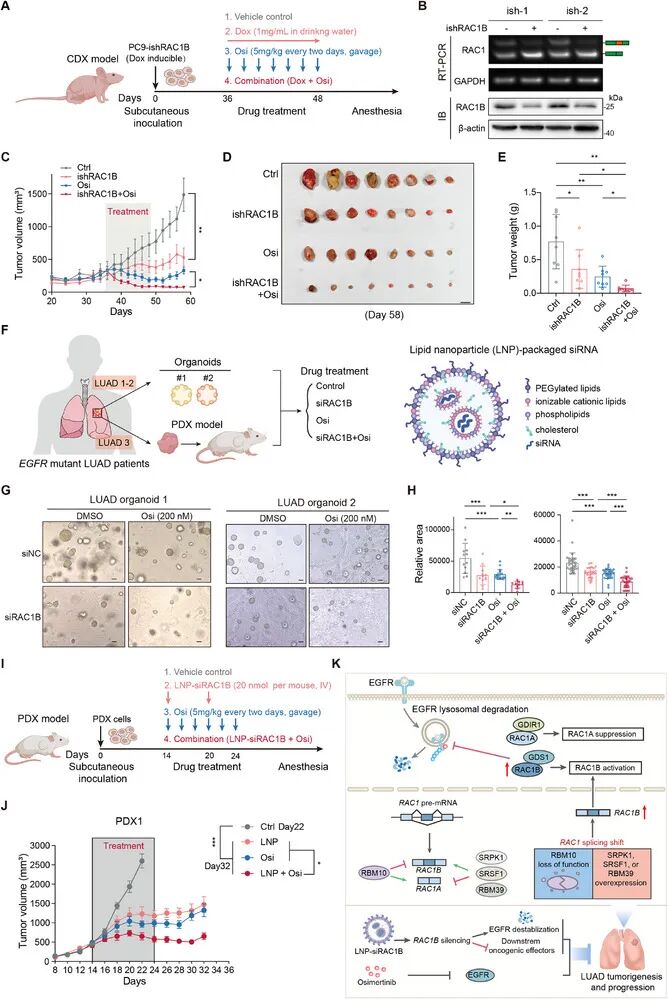
图4. RAC1B促癌机制及治疗策略
和元助力
和元生物有幸为客户提供GDIR1和GDS1过表达慢病毒,用于感染PC9(人肺腺癌细胞)
研究四
RANKL/PD-1 双靶向阻断对伴有 KRAS 突变的晚期肺腺癌患者显示出生存获益
文章标题:RANKL/PD-1 dual blockade demonstrates survival benefit for patients with advanced lung adenocarcinoma harboring KRAS mutations
发表期刊:Cell Reports Medicine(IF 11.7)
研究团队:中国医学科学院北京协和医学院
研究背景:KRAS突变的晚期肺腺癌(LUAD)患者对免疫检查点抑制剂(如PD-1/PD-L1抑制剂)响应有限,耐药机制尚不明确。研究表明RANKL信号通路可能参与免疫抑制微环境(TIME)的调控,但其具体机制及治疗潜力亟待探索。
结果:研究者首先通过NCC队列分析发现,接受RANKL抑制剂联合免疫检查点抑制剂(RLICi)治疗的KRAS突变LUAD患者相比非RLICi组具有更长的无进展生存期和更高的持久临床缓解率。进一步通过生物信息学分析多个公共数据库(TCGA、GEO、CPTAC数据库)和IHC验证(CICAMS队列),确认KRAS突变LUAD中RANKL显著高表达,且与PD-L1低表达、CD8⁺ T细胞浸润减少显著负相关。为明确RANKL调控PD-L1的机制,研究者构建了RANKL过表达的LUAD细胞系(H358、A427、LLC),发现RANKL过表达通过激活PI3K-AKT通路抑制PD-L1表达,并通过抑制巨噬细胞趋化功能及CXCL9/10/11的分泌,削弱CD8+ T细胞招募。后续作者建立了小鼠皮下移植瘤模型(LLC和CMT167细胞),发现RLICI联合治疗显著抑制肿瘤生长,并促进CD8+ T细胞和M1巨噬细胞浸润。最后前瞻性DEMAIN试验显示:接受RLICi维持治疗的KRAS突变LUAD患者mPFS达347天,且安全性良好,未发生严重不良事件,提示RANKL可作为预测RLICi疗效的潜在生物标志物。
总结:本研究首次在机制上阐明RANKL通过PI3K-AKT通路下调PD-L1表达并抑制巨噬细胞功能,从而驱动KRAS突变LUAD的免疫逃逸。通过临床前及临床数据验证了RLICi联合治疗的协同抗肿瘤效应,为该人群提供了新的治疗策略。研究结果支持RANKL作为预测免疫治疗响应的生物标志物及潜在治疗靶点。
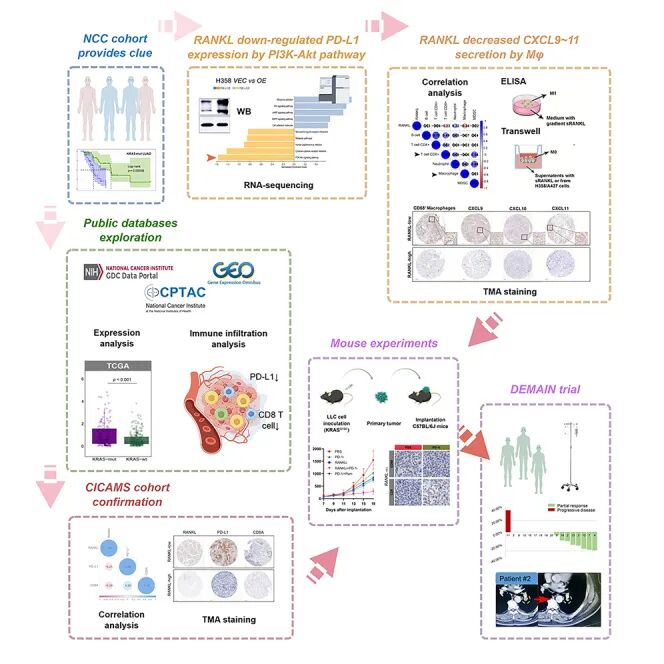
图5. 示意图
和元助力
和元生物有幸为客户提供RANKL过表达慢病毒和si-RANKL,用于感染NCI-H358(人非小细胞肺癌细胞)、A427(人肺癌细胞)、NCI-H322(人支气管肺泡癌细胞)、CMT167和LLC(小鼠肺癌细胞)。
研究五
GABA 通过调节代谢重编程介导非小细胞肺癌脑转移的发生发展
文章标题:GABA regulates metabolic reprogramming to mediate the development of brain metastasis in non-small cell lung cancer
发表期刊:Journal of Experimental & Clinical Cancer Research(IF 11.4)
研究团队:同济大学医学院附属上海肺科医院
研究背景:脑转移(BrM)对非小细胞肺癌(NSCLC)患者的预后和生活质量构成重大挑战。GABA是中枢神经系统(CNS)中的抑制性神经递质,已被认为与多种肿瘤的进展有关。然而,其在 NSCLC 脑转移中的潜在作用及其潜在机制尚不明确。
结果:研究团队首先通过左心室注射筛选并构建了H460BrM和PC9BrM 脑转移细胞系,并通过多组学分析发现这些细胞中GABA水平显著升高,而其降解酶ABAT表达下降。体外实验表明,外源性GABA以剂量依赖性方式增强NSCLC细胞的增殖、迁移和侵袭能力。体内左心室注射模型进一步证实,GABA处理组小鼠的脑转移发生率显著提高。机制研究发现,脑转移倾向的肿瘤细胞通过下调FOXA2抑制ABAT表达,导致GABA积累,进而激活NF-κB通路。最后通过IHC与共培养实验发现,脑转移灶周围星形胶质细胞(GFAP+)富集,GABA 可促进星形胶质细胞迁移并激活其 NLRP3 炎症小体,激活的星形胶质细胞在低营养环境下(1% FBS)更能促进肿瘤细胞增殖,而 MCC950 抑制 NLRP3 后该效应消失,证实肿瘤细胞与星形胶质细胞通过 GABA 形成正反馈 loop,构建促转移微环境。
总结:本研究首次系统揭示了GABA通过FOXA2/ABAT/GABA轴激活NF-κB通路,促进NSCLC脑转移的新机制,并阐明了肿瘤细胞通过模拟神经元活动“驯化”星形胶质细胞的微环境重塑策略。该研究为GABA作为脑转移治疗靶点提供了理论基础和实验依据。
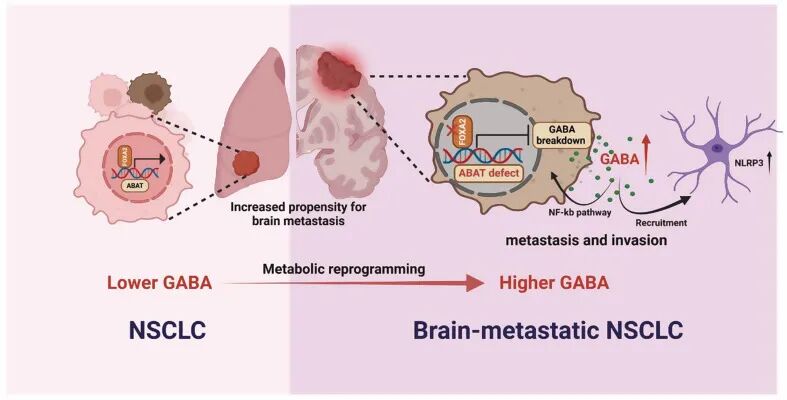
图6. 机制示意图
和元助力
和元生物有幸为客户提供ABAT过表达慢病毒和si-FOXA2,用于感染H460(人大细胞肺癌细胞)。
研究六
CDKN2A缺失可增强携带EGFR突变非小细胞肺癌的免疫治疗疗效
文章标题:Loss of CDKN2A Enhances the Efficacy of Immunotherapy in EGFR Mutant Non-Small Cell Lung Cancer
发表期刊:Cancer Research(IF 12.5)
研究团队:沈阳药科大学
研究背景:EGFR突变是非小细胞肺癌(NSCLC)的常见驱动突变,但这类患者通常对免疫检查点抑制剂(ICIs)反应不佳。然而,部分EGFR突变患者仍能从免疫治疗中获益,提示共突变基因可能影响免疫疗效。本研究旨在探索EGFR共突变对免疫治疗的影响及其机制。
结果:研究团队首先通过 cBioPortal 数据库系统分析EGFR突变型非小细胞肺癌(NSCLC)的共突变谱,筛选出 7 种高频共突变基因(失活突变:TP53、CDKN2A、RB1; 功能获得突变:NKX2-1、PIK3CA、MDM2、CDK4)。接着作者利用 Tet-on 诱导系统和 CRISPR/Cas9 技术在人 PC9-Em 和小鼠 LLC-Em 细胞中构建7种相应共突变模型。通过肿瘤细胞-免疫细胞共培养实验发现,EGFR/CDKN2A和EGFR/TP53共突变细胞对PD-1单抗治疗最为敏感,IFN-γ分泌水平显著升高,而EGFR/MDM2共突变细胞则表现出相对耐药性。进一步在小鼠模型中证实,EGFR/CDKN2A共突变肿瘤对EGFR-TKI联合PD-1抗体治疗响应最佳,肿瘤抑制率达75.1%,且CD8+ T细胞浸润明显增加。
为明确 EGFR/CDKN2A 共突变增强免疫治疗敏感性的机制,研究团队借助 TISIDB 数据库和 LUAD 蛋白质组学数据发现,CDKN2A 突变患者中 PD-L2 表达显著升高,且 CDKN2A 蛋白与 PD-L2 蛋白呈负相关,临床数据进一步显示高 PD-L2 表达的 NSCLC 患者对 PD-1 治疗响应更好。后续的机制研究表明CDKN2A缺失通过减少c-Myc的泛素化降解来上调PD-L2表达。此外,EGFR突变通过激活MAPK通路进一步增强了c-Myc和PD-L2的表达,形成协同效应。
在治疗策略方面,PD-L2阻断抗体在体外共培养实验中显著增加EGFR/CDKN2A共突变细胞的凋亡率,并促进CD8+ T细胞介导的肿瘤杀伤作用。使用CD8中和抗体后,这种效应被显著削弱,证实PD-L2阻断的作用依赖于CD8+ T细胞。最后作者筛选出一种天然小分子化合物锌十一烯酸(ZU),能有效抑制PD-L2/PD-1结合。实验表明,ZU在免疫细胞存在下选择性抑制EGFR/CDKN2A共突变肿瘤细胞,并与EGFR-TKI联用显著增强抗肿瘤效果,重塑TIME,促进CD8+ T细胞活化和浸润。
总结:该研究首次明确 EGFR/CDKN2A 共突变可作为NSCLC免疫治疗的新型生物标志物,同时揭示 “CDKN2A-p14ARF-c-Myc-PD-L2” 调控轴及 EGFR-MAPK 通路的协同作用机制,该研究不仅为EGFR突变型NSCLC的免疫治疗"禁区"提供了突破性解决方案,还开发了具有转化潜力的新型小分子免疫治疗药物。
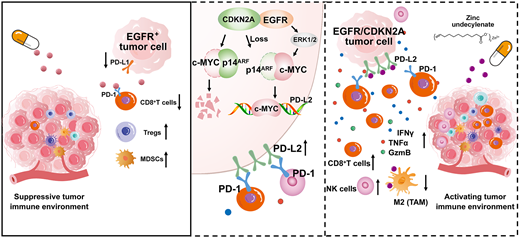
图7.机制示意图
和元助力
和元生物有幸为客户提供CDKN2A、TP53、RB1敲除慢病毒和PD-L2过表达慢病毒,用于感染LLC-Em(携带EGFR外显子19缺失突变的小鼠肺癌细胞)和PC9-Em(携带EGFR外显子19缺失突变的人肺腺癌细胞)
和元服务
和元一直致力为肿瘤研究提供整体解决方案,从基因筛选、基因功能研究到机制研究,提供质粒构建、病毒包装、基因过表达、基因干扰、基因敲除稳转株构建、细胞功能学实验、双荧光素酶检测服务到动物模型构建一站式技术服务!
业务咨询:15800353038

