- 移动端


上海和元生物技术(集团)股份有限公司品牌商
14 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5

推荐产品
公司新闻/正文
Cancer Cell|姚兵/向俊研究团队揭示靶向HIF-1α-MT2A轴可克服肿瘤细胞的铜死亡抗性
983 人阅读发布时间:2025-09-15 16:21
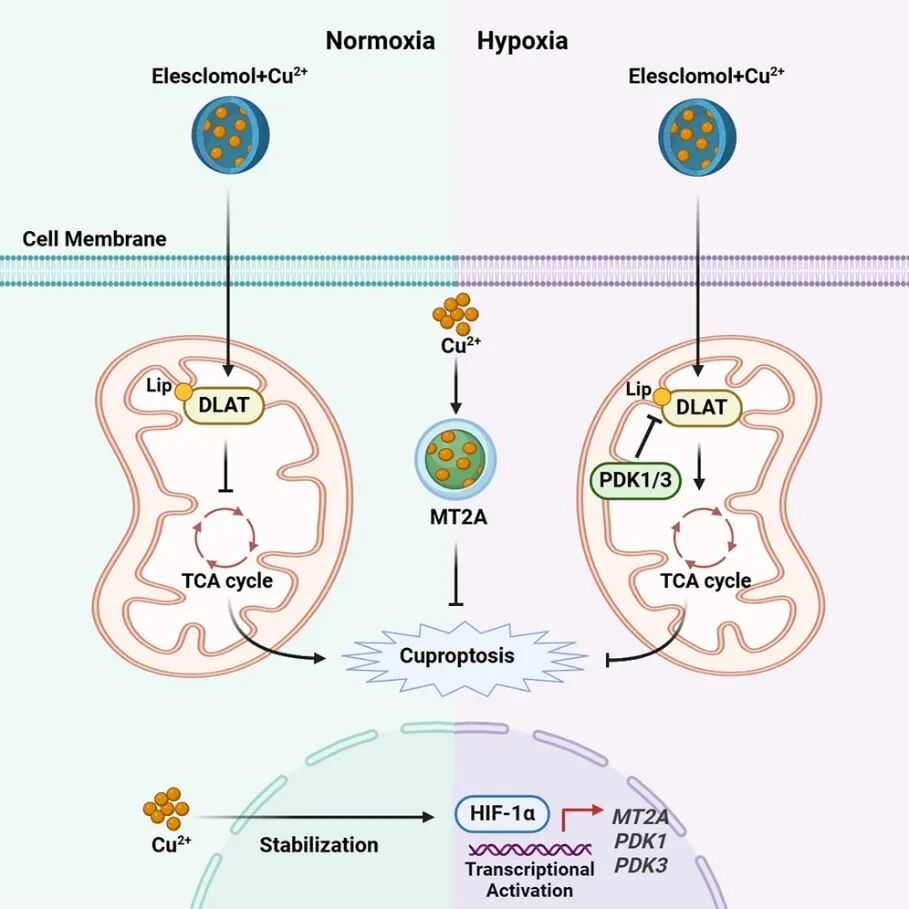
众所周知,多细胞生物在发育过程中,存在着多种预定的、受到精确控制的细胞程序性死亡,如细胞凋亡(Apoptosis)、坏死性凋亡(Necroptosis)、细胞焦亡(Pyroptosis),以及铁死亡(Ferroptosis)等。铜死亡(Cuprotosis)是Tsvetkov等人于2022年在Science上首次报道的一种新的死亡方式,其特点是铜离子载体促进铜离子进入细胞,导致脂酰化蛋白的异常聚集、线粒体呼吸中铁硫簇蛋白的破坏以及蛋白质毒性相关的应激反应的发生,最终导致细胞死亡。铜死亡作为新近发现的细胞死亡方式,在肿瘤治疗中具有重大潜力。
2025年3月6日,南京医科大学基础医学院姚兵、复旦大学附属肿瘤医院向俊、复旦大学上海医学院周祥、复旦大学附属肿瘤医院王宇、南京医科大学第一附属医院(江苏省人民医院)秦超、南京明基医院/南京医科大学基础医学院陈云等作为共同通讯作者,在 Cancer Cell 期刊发表了题为:Hypoxia inducible factor-1α drives cancer resistance to cuproptosis 的研究论文。通过空间转录组测序(ST-seq)、单细胞RNA测序(scRNA-seq)发现缺氧区域与铜死亡抵抗细胞簇高度重叠,进一步HIF-1α通过激活PDK1/3抑制DLAT表达,减少铜在三羧酸循环中的积累,同时促进MT2A表达螯合铜离子,从而保护肿瘤细胞免受铜毒性的影响,最后揭示铜能够稳定HIF-1α蛋白,形成正反馈机制。
研究结果
1. HIF-1α 在缺氧肿瘤微环境中驱动对铜死亡的抗性
为了研究缺氧在铜死亡中的作用,对透明细胞肾细胞癌(ccRCC)和乳头状甲状腺癌(PTC)进行了空间转录组测序(ST-seq)。在缺氧评分和铜死亡评分之间观察到负相关,缺氧的肿瘤微环境区域显示出较低的铜死亡评分(图1A-B)。在 PTC 的单细胞水平上(scRNA-seq)验证了缺氧与铜死亡之间的关联,缺氧细胞簇与抗铜死亡细胞簇高度重叠,且缺氧评分与铜死亡评分呈负相关(图 1C-D)。
缺氧显著减轻了 ES +Cu 刺激诱导的癌细胞的铜死亡(图 1E)。为了确定 HIF-1α 是否参与缺氧驱动的铜死亡抗性,分别在 TPC1 和 HCT116 细胞中敲低或过表达 HIF-1α。有趣的是,敲低 HIF-1α 显著恢复了癌细胞在缺氧条件下对 ES+Cu 诱导的铜死亡的敏感性。过表达 HIF-1α 在常氧和缺氧条件下均促进了对铜死亡的抗性。综上所述,HIF-1α控制着肿瘤中缺氧诱导的铜死亡抗性。
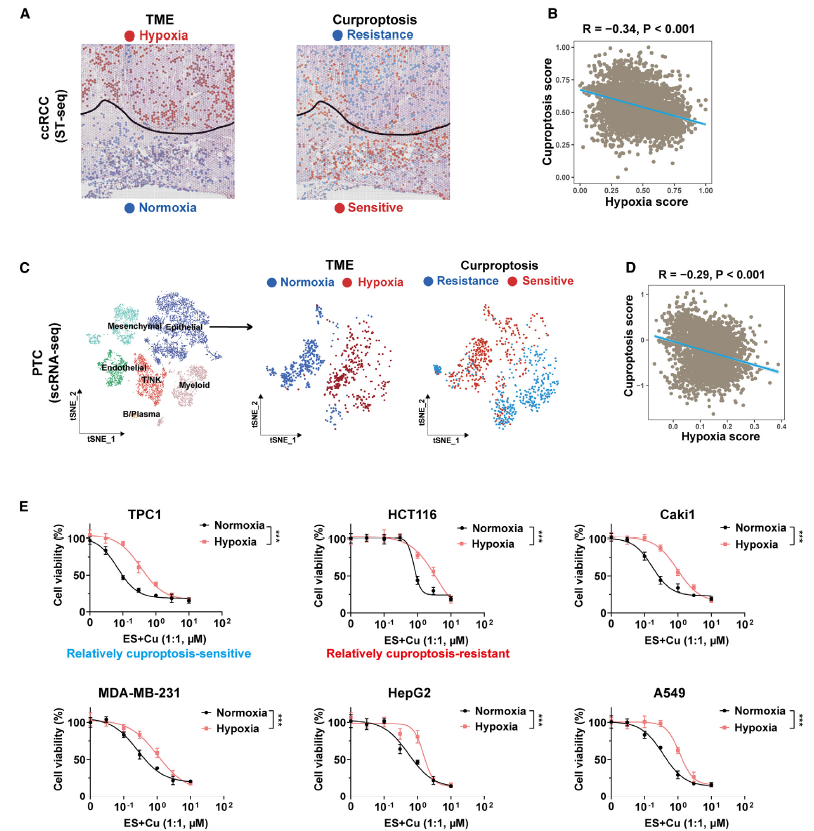
图1 HIF-1α 在缺氧的肿瘤微环境中驱动对铜死亡的抗性
2.HIF-1α 抑制铜靶蛋白 DLAT 的表达
先前研究表明,ES+Cu靶向三羧酸(TCA)循环中的脂酰化蛋白(DLAT 和 DLST),从而抑制线粒体呼吸(图 2A)。值得注意的是,缺氧条件下 DLAT 和脂酰化 DLAT(lip-DLAT)均显著下调(图 2B)。DLAT 和 lip-DLAT 的水平与氧浓度呈正相关(图 2C)。此外,在 HIF-1α 敲低的细胞中,DLAT 和 lip-DLAT 的下调现象消失(图 2D)。这些结果表明缺氧诱导的 DLAT 和 lip-DLAT 抑制具有 HIF-1α 依赖性。
此外,证实了在PTC和 CRC中,HIF-1α 与 DLAT 之间存在负相关(图 2F)。DLAT 是 PDH 复合物的一个亚基,缺氧和ES+Cu处理均降低了 PDH 的活性,而 HIF-1α 的敲低则使其恢复(图 2G)。DLAT 的过表达不会影响短期细胞活力,但能恢复缺氧条件下细胞对 ES+Cu 的敏感性(图 2H-I)。这些结果表明,DLAT 介导了缺氧诱导的、HIF-1α 调节的铜死亡过程。
先前研究证实,丙酮酸脱氢酶激酶(PDK1/2/3/4)是 HIF-1α 的直接转录靶点。RNA-seq及WB验证,发现缺氧诱导的 PDK1 和 PDK3 上调是 HIF-1α 依赖性的(图 2J-K)。敲低 PDK1 和 PDK3 恢复了癌细胞中 DLAT 的表达以及 ES+Cu诱导的铜死亡(图 2L-M)。此外,构建了 PDK1 和 PDK3 双敲除(DKO)细胞,DKO 细胞中 DLAT 的表达增加更为显著。然而,在 DKO 细胞中,缺氧无法降低 DLAT 的表达,这表明 PDK1/3 对于 HIF-1α 调节的 DLAT 抑制至关重要。还研究了 PDHA1 的磷酸化情况,并证实敲低 HIF-1α 或 PDK1/3 会抑制 p-PDHA1(图 2K 和 2L)。
鉴于 PDKs 通过PDHA1 和 PDHA2 的磷酸化来调节葡萄糖和脂肪酸代谢。采用磷酸化蛋白质组在 DLAT 中鉴定出一个磷酸化位点(S100),该位点可被 sgPDK1 下调。特定位点的磷酸化可能会促进泛素连接酶的识别,从而导致泛素介导的降解。敲低 PDK1 和 PKD3 均抑制了 DLAT 的 Ub 和 p-Ser 水平。此外,敲低 HIF-1α 降低了 DLAT 的 Ub 和 p-Ser 水平,而在 HIF-1α 敲低的细胞中过表达 PDK1 和 PDK3 则恢复了 DLAT 的 Ub 和 p-Ser 水平。总体而言,HIF-1α 转录激活 PDK1 和 PDK3,从而抑制 DLAT 表达,降低 PDH 活性,并促进 p-PDHA1 的积累。
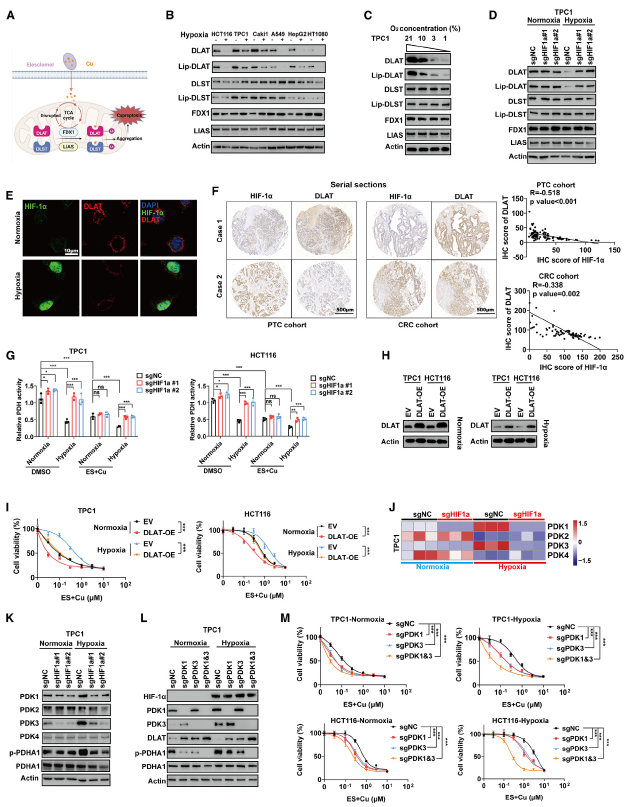
图2 HIF-1α 抑制铜靶蛋白 DLAT 的表达
3.铜稳定 HIF-1α 蛋白
采用代谢组分析了缺氧与铜死亡之间的相互作用。ES+Cu刺激抑制了多种细胞活动以诱导铜死亡,导致了糖酵解相关代谢物(如乳酸和丙酮酸)的积累,并增加了细胞外酸化率(ECAR)和葡萄糖摄取,这表明糖酵解被激活(图 3A)。这些反应也是 HIF-1α 依赖性的(图 3B-C)。引人注目的是,亚致死剂量的 ES+Cu在常氧和缺氧条件下均促进了细胞核中 HIF-1α 的表达(图 3D)。
通过用蛋白质合成抑制剂 CHX 处理,检测到在 ES+Cu处理的细胞中 HIF-1α 蛋白的稳定性显著增加(图 3E)。ES+Cu处理显著降低了 HIF-1α 的泛素化(图 3F)。金属亲和力测定表明,HIF-1α 对负载铜的树脂的亲和力比对其他金属更强,表明它们之间存在潜在的相互作用(图 3G)。分子对接也揭示了 HIF-1α 上几个高可信度的铜结合位点(图 3H)。微尺度热泳(MST)测定证实CuCl2与 HIF-1α 结合的平衡解离常数(KD)为 20.6 ± 4.6 μM(图 3I)。
随后,检测了几种受 HIF-1α 靶向的糖酵解酶,包括 Glut1(SLC2A1)、HK2、PKM2、GAPDH 和 LDHA。ES+Cu 刺激增强了这些酶的表达,在缺氧条件下 Glut1 的反应最为明显(图 3J)。敲低 Glut1 在常氧和缺氧条件下均显著增强了 ES+Cu 诱导的铜死亡(图 3K)。这些数据共同表明,HIF-1α 激活的糖酵解是细胞在铜死亡应激下的一种适应性反应和替代能量补充方式。
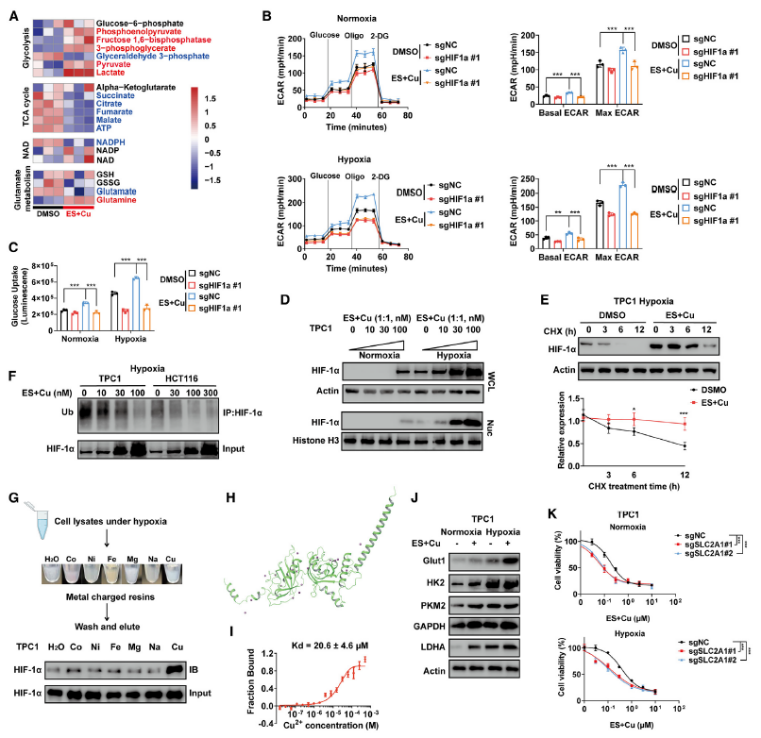
图3 铜稳定 HIF-1α 蛋白
4.HIF-1α 在缺氧条件下促进金属硫蛋白的积累
铜的转运和沉积在铜死亡诱导中也起着关键作用。在分析 HIF-1α 敲低的 TPC1在常氧和缺氧条件下的转录谱时,发现金属硫蛋白(MT)家族成员,如MT2A、MT1L、MT1X 和 MT1F,在缺氧条件下上调,在 HIF-1α 敲低的 TPC1 细胞中下调(图 4A)。此外,MTs,特别是 MT2A、MT1L、MT1X 和 MT1F,在 ES+Cu 刺激后显著上调(图 4B)。
有趣的是,MT2A 的蛋白质表达在缺氧条件下均显著上调(图 4C)。免疫荧光染色显示,MT2A 在细胞核和细胞膜中呈现缺氧特异性的聚集,在常氧条件下则减少(图 4D)。此外,在 TPC1 细胞中,MTs 的表达随着氧浓度的降低而增加(图 4E-F)。HIF-1α 敲除抑制了缺氧条件下 MTs 的表达(图 4G-H)。相反,ES+Cu刺激上调了 MTs 的表达(图 4I-J)。ES+Cu协同增强 MTs 的表达(图 4K)。MST 分析显示MT2A 与铜特异性结合(图 4L)。
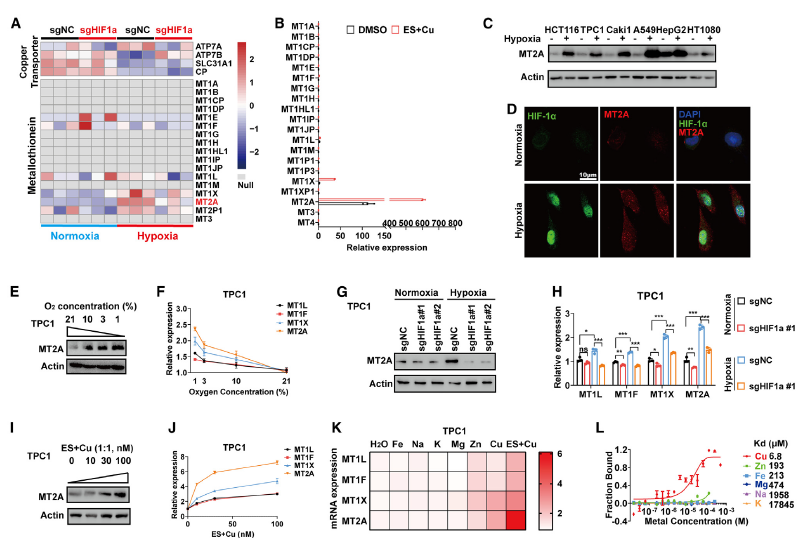
图4 HIF-1α在缺氧条件下促进金属硫蛋白的积累
5.MT2A 螯合线粒体铜并导致铜死亡抗性
在 PTC(单细胞 RNA 测序和空间转录组测序)和 ccRCC(空间转录组测序)中,过表达 MT2A 的肿瘤细胞具有铜代谢途径激活的特征(图 5A)。MT2A 主要存在于细胞核和细胞膜中,与线粒体的共定位极少(图 5B)。在常氧或缺氧条件下从 TPC1 细胞分离的线粒体中也未检测到 MT2A(图 5C)。由于线粒体是 ES+Cu 诱导铜死亡的主要靶点,且 MT2A 缺乏线粒体定位,因此推测线粒体铜水平可能区分 MT2A 螯合的铜和驱动铜死亡的 “游离” 铜。正如预期,缺氧条件下细胞线粒体铜水平显著低于常氧条件(图 5D)。线粒体铜含量随着氧水平的下降而逐渐降低(图 5F),但在缺氧条件下 HIF-1α 缺失的细胞中恢复(图 5G)。
由于 MT2A 是表达量最高的 MT,并且对缺氧和 ES-Cu 刺激的反应最强,接下来通过过表达和敲低 MT2A 来研究其具体作用。在 HIF-1α 缺失的细胞中过表达 MT2A 可降低线粒体铜含量,并在常氧和缺氧条件下恢复细胞活力,而敲低 MT2A 则会升高线粒体铜含量并加剧 ES+Cu 诱导的铜死亡(图 5H-J)。
为了确定 HIF-1α 是否直接调节 MT2A 的表达,进行了染色质免疫沉淀ChIP-qPCR 分析。在缺氧条件下,MT2A 启动子在 HIF1α 免疫沉淀复合物中显著富集,过表达 HIF-1α 进一步促进了这种富集(图 5K)。鉴于 HIF-1α 主要识别其靶基因启动子中的缺氧反应元件(HRE)(5'-RCGTG-3'),分析了 MT2A 的启动子区域,发现了 2 个潜在的 HRE。缺氧显著促进了野生型 MT2A 启动子的荧光素酶活性。将 HRE 的核心共有基序(CGT)突变为 5'-TAC-3' 后,荧光素酶活性无法检测到(图 5L-N)。最后,证实了HIF-1α 和 MT2A 之间存在显著的正相关(图 5O)。综上所述,HIF-1α 促进 MT2A 的积累以捕获线粒体铜,从而保护癌细胞免受铜毒性的影响(图 5P)。
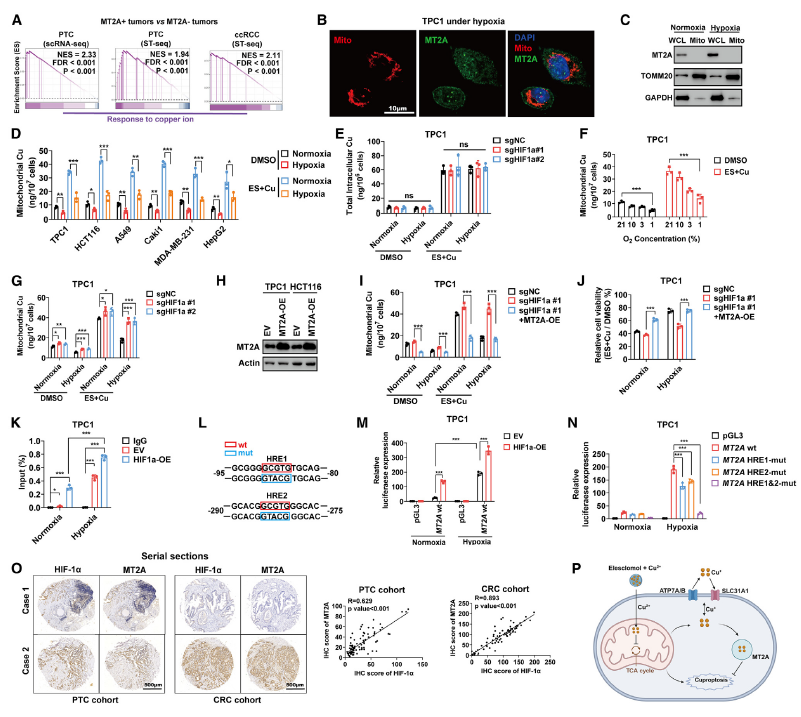
图5 MT2A 螯合线粒体铜并导致对铜死亡的抗性
6.MT2A 促进癌细胞在铜应激下的长期增殖
在TCGA中,MT2A高表达预示着多种癌症患者的生存率更低、肿瘤分期更晚(图 6A)。同样,在CRC、ccRCC 和 PTC 队列中,较高的 MT2A 表达也与较差的生存率和晚期肿瘤分期相关(图 6B-C)。这些发现强调了缺氧相关途径与癌症预后不良之间的潜在联系。
敲低 MT2A后,这些细胞在培养 15 天后失去了集落形成能力(图 6D)。在长期培养且不额外补充 ES+Cu 的情况下,MT2A 缺失的细胞中线粒体铜积累显著增加(图 6E)。用铜螯合剂 TTM 处理可有效降低线粒体铜积累,并恢复 MT2A 缺失细胞受损的克隆形成能力(图 6D-E)。通过敲低 SLC31A1 对铜转运进行抑制,在 MT2A 缺失的细胞中也显示出挽救效果(图 6D-E)。此外,体内实验表明,敲低 MT2A 适度抑制了肿瘤生长,而 TTM 处理可完全逆转这种抑制作用(图 6F-H)。这些研究结果突出了 MT2A 在减轻铜诱导的细胞毒性方面的关键作用,从而支持癌细胞在生理铜条件下的存活和增殖。
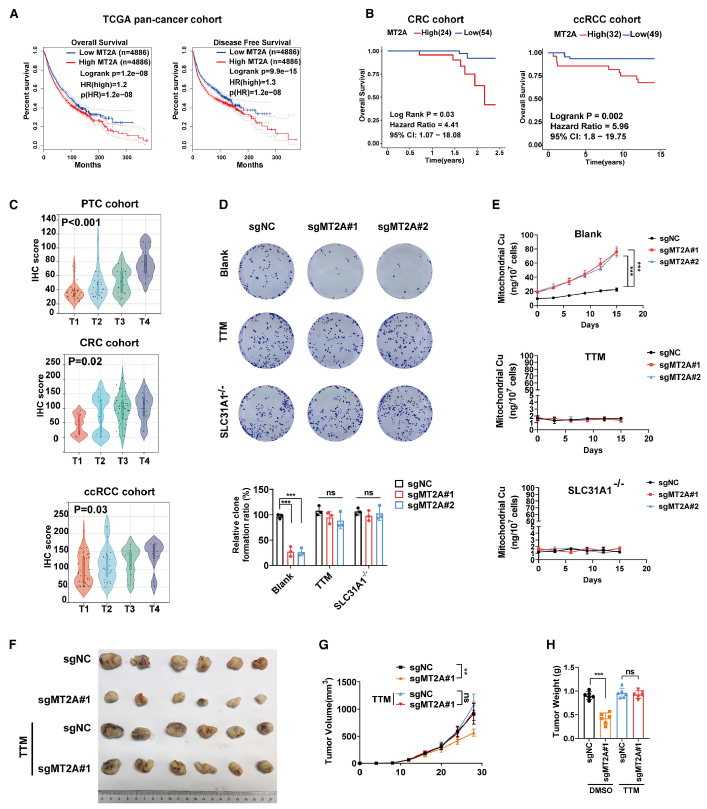
图6 MT2A 促进癌细胞在铜应激下的长期增殖
7.HIF-1α 抑制剂使肿瘤细胞对铜死亡敏感
为了确定 ES+Cu 处理在体内是否也能抑制肿瘤生长,体内实验分析了ES+ Cu的治疗效果。ES+ Cu联合刺激显著抑制了肿瘤生长(图 7A-B)。除了在肿瘤中的积累外,还观察到肝脏中 MT2A 水平显著增加(图 7C)。随着铜剂量从 0.25mg/kg 增加到 2.5mg/kg,ES+Cu 的肿瘤抑制效果增强(图 7D-E)。此外,ES+Cu(2.5mg/kg)显著上调了肿瘤和肝脏中 MT2A 的表达(图 7F)。
PX-478 是一种已知的化合物,可阻止 HIF-1α 的去泛素化并增强其蛋白酶体依赖性降解。在体外缺氧条件下,PX-478 与 ES+Cu 联合使用对抑制细胞增殖表现出有趣的协同作用,并且这种作用可被铜螯合剂 TTM 逆转。PX-478 可有效促进 ES+Cu 诱导的铜死亡(图 7G-I)。此外,在 AOM/DSS 诱导的同基因小鼠 CRC 模型中研究了联合疗法的有效性和安全性(图 7J)。与细胞系来源的异种移植(CDX)模型一致,PX-478 和 ES+Cu 在体内肿瘤微环境中也协同抑制了 CRC 的肿瘤生长,且没有肝脏相关的副作用(图 7K-N)。考虑到对 PX-478 特异性的担忧,还使用腺相关病毒(AAV)靶向 HIF-1α。与 PX-478 的结果类似,AAV-shHIF1A 与低剂量 ES+Cu(0.25mg/kg)联合使用也表现出显著的协同作用,并强烈抑制肿瘤生长。总的来说,HIF-1α抑制剂PX-478可以增强ES+Cu联合治疗的抗肿瘤效果。在异种移植瘤模型和同位CRC模型中,PX-478联合ES+Cu治疗均可显著抑制肿瘤生长,降低肿瘤组织中MT2A的表达水平。这些结果表明,靶向HIF-1α-MT2A轴可以克服肿瘤细胞的铜死亡抗性,为开发新的癌症治疗策略提供了重要的实验依据。
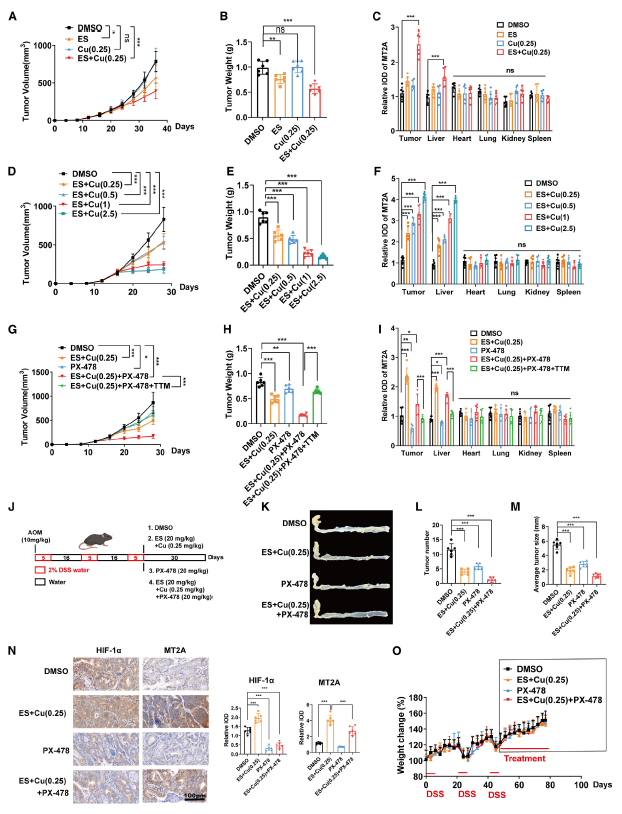
图7 HIF-1α 抑制剂使肿瘤细胞对铜死亡敏感
小 结
HIF-1α是肿瘤耐受铜毒性的关键驱动因素;而涉及到的细胞类型和表型为缺氧条件下的肿瘤细胞,分子信号轴为HIF-1α-PDK1/3-DLAT和HIF-1α-MT2A。靶向HIF-1α-MT2A轴可以克服肿瘤细胞的铜死亡抗性,为开发新的癌症治疗策略提供了重要的实验依据。所涉及的组学技术包括:空间转录组测序(ST-seq)、单细胞RNA测序(scRNA-seq)、转录组测序(RNA-seq)、磷酸化蛋白组(Phosphoproteomics)、代谢组(LC-MS/MS)、染色质免疫沉淀ChIP-qPCR 分析等。
和元服务
和元生物提供多组学服务(如转录组、Astral蛋白组、非靶向代谢组、表观组和微生物组等)和时空组学服务,如单细胞转录组测序(10x、墨卓等多平台)和空间转录组测序(10x CytAssist、10x Visium HD),助力您的科研!
业务咨询:15800353038

